12月上半月主要在做实验,文献阅读有一些耽搁,下个半月坚持一下。
2021年12月22日
scDeepSort: a pre-trained cell-type annotation method for single-cell transcriptomics using deep learning with a weighted graph neural network
2021 NAR 一篇比较新的文章,核心是通过在大规模数据上进行所谓的“预训练”,从而对新数据集上的细胞类型进行标注。整个工作的基础是建立在HCL和MCA数据集上的,作者开源了实验和测试代码:https://github.com/ZJUFanLab/scDeepSort。作者一共使用了764,741个细胞进行训练,一共有56个人类和32个小鼠的组织,之后作者和SingleR (8), CHETAH (9), scMap (10), scID (11), scPred (12), ACTINN (13), CellAssign (14), Garnett (15), SCINA (16), singleCellNet (17) and support vector machine (SVM)等方法在76个测试数据集上一共265,489个细胞上进行比较,他们的模型是SOTA的并达到了83.79%的准确率。
根据题目知道,他们需要构建的是一个图卷积网络,那么首先要构建出图的结构和节点的特征。他们将细胞和基因都作为图的节点,将细胞i在基因j上的表达作为i,j之间的边权重,而基因的特征使用了PCA的前200维度,而细胞的特征是基因特征的加权平均,这里作者没有特别说明权重是什么,我感觉还是边的权值。然后在这张图的基础上作者使用了一层图卷积网络,借鉴了GraphSAGE的思想,目的是为了得到每个节点的embedding向量,然后作者再用这个向量经过一层MLP来进行分类任务。训练的时候作者就先考虑已有数据的图,然后在测试时,先把测试数据接在已有数据的图上,边的权值是在测试数据集上的基因表达值,然后再用已经训好的GNN求得每个测试细胞的embedding,然后再进行分类,中间图卷积的公式是,其中h 为节点特征,k表示网络的当前层数,$\alpha$是节点selfloop的权重,$\beta$是边之间的权重:
$$
h_{i}^{k}=\sigma\left(W^{k-1} \frac{\alpha h_{i}^{k-1}+\sum_{j \in N(i)} \beta_{j} a_{i j} h_{j}^{k-1}}{1+|N(i)|}+b^{k-1}\right)
$$
也就是说,这里的边只有基因和细胞存在联系,作者其实没有考虑细胞和细胞之间的关系,即使图卷积了一层,也只是考虑了一阶邻居的影响。这张图更能说明作者的想法:
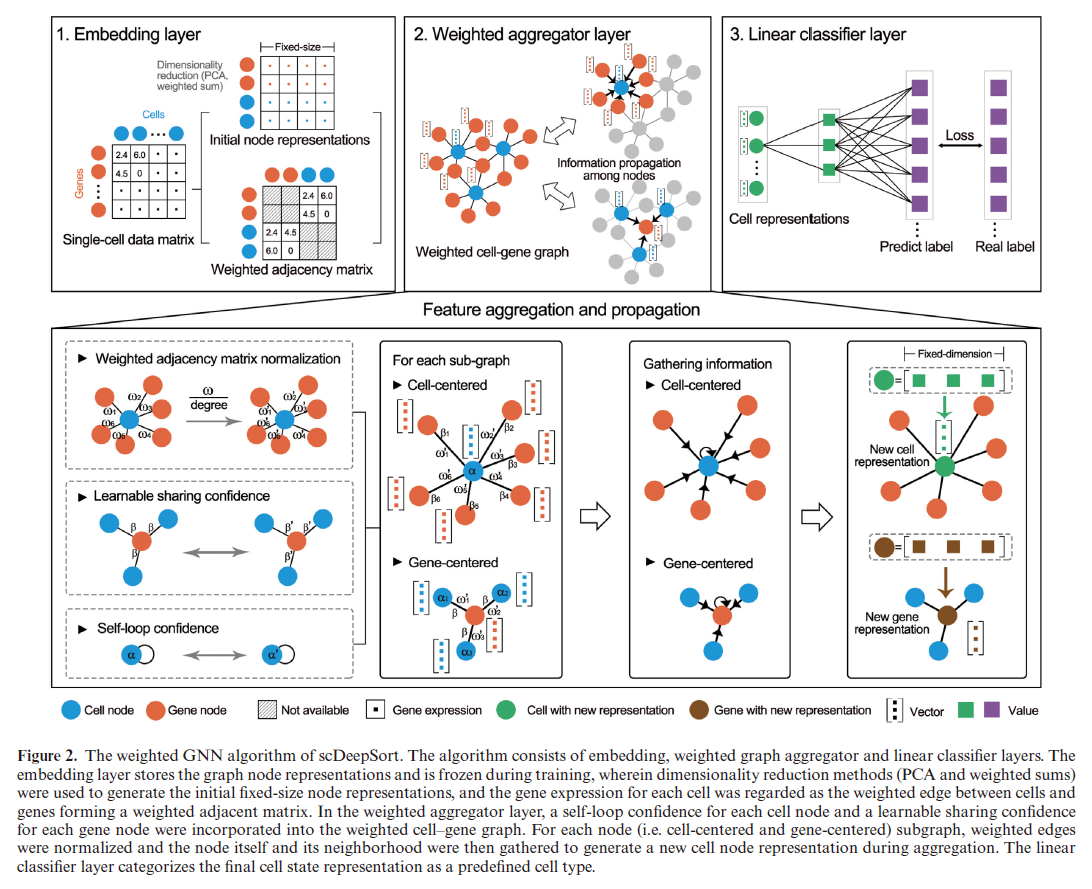
思路讲清楚了,更多的测试结果我就在这里不放了,结论就是比之前的方法都要好一些。在讨论部分,作者还用了像是GNNExplainer这样的工具对图卷积的预测过程进行了可视化,发现在区分巨噬细胞时,确实找到了几个基因是众所周知的Marker(但是本来巨噬细胞就表达这些基因,所以他在图上的连接强度高,这不也是很正常的吗?)。总的来说是值得一看的文章。
2021年12月23日
scGCN is a graph convolutional networks algorithm for knowledge transfer in single cell omics
Nature Communication 2021 这篇文章建议和“DSTG: deconvoluting spatial transcriptomics data through graph-based artificial intelligence”一起读,属于是一虾两吃了。文章的思路很简单,就是把两个单细胞数据通过某些度量的计算构建出一张近邻图,其中图的节点是每个细胞,而图的边就是节点之间的相似度。然后,整个图作为图卷积网络的输入进行训练,与上一篇文章的模型不同,这里每个细胞是和细胞直接相连的,没有通过基因作为连接,所以他必须在训练时就把测试数据放进来。图卷积网络的输出就是每个节点的细胞类型归属,而在DSTG文章里,图卷积的输出就是每个Spot细胞类型的可能性比例。而这个输出是不分训练数据和测试数据的,所以模型就可以根据reference数据的邻边关系计算出query细胞的细胞类型。但说实话,相比于scDeepsort,这篇文章确实差了一些。
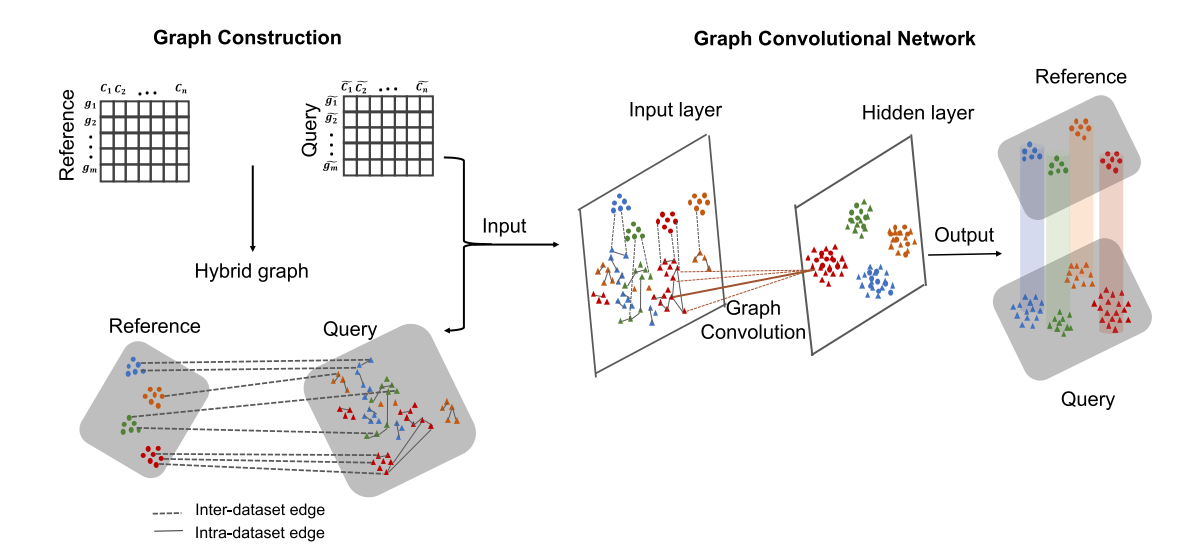
2021年12月23日
Representation Learning and Translation between the Mouse and Human Brain using a Deep Transformer Architecture
ICML2020 Workshop 总体上是一个计算不够强,生物味道也不足的文章,但是他是一个用了transformer结构的工作,他想解决的问题是想把两组在相似组织得到的细胞类型进行merge,本质上属于去除batch effect的工作。文章的图还是挺迷惑的:
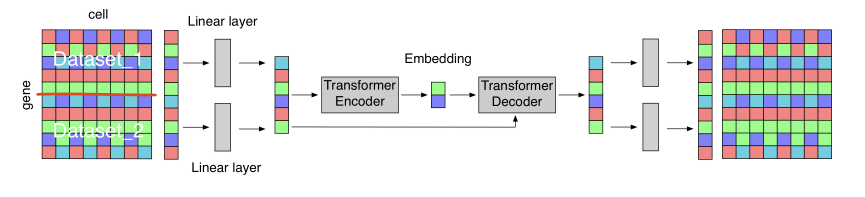
具体来说,对于两个基因表达数据$X_{A} \in \mathbb{R}^{a \times n}$和$X_{B} \in \mathbb{R}^{b \times m}$,作者先利用线性层得到了与数据数据相同大小的表达矩阵,线性层的参数是$1 \times n$和$1 \times m$的向量,然后把这个向量分别扩展a列和b列,用来表示数据batch的信息。之后,Transformer的输入就是batch信息加上经过变换后的表达矩阵。transformer会在中间生成embedding,这是作者之后重点比较的内容,整个模型用重建损失作为Loss进行优化。呃但作者中间的符号很奇怪,有一些矩阵按照作者的写法是根本没法计算的。
在结果部分,作者用1000个细胞的embedding做可视化,基本上就是发现两类细胞都有相同的流形,其中细胞类型在流形上的分布也非常一致,呃但从作图以及结果的分析上看,其实结论也不是很有趣。
2021年12月24日
Gene Transformer: Transformers for the Gene Expression-based Classification of Lung Cancer Subtypes
arxiv 2021 利用Transformer框架做细胞类型分类的工作,作者认为文章的主要卖点是利用Transformer的结构可以不需要预先的特征筛选。但是实际上对于单细胞来说,基因数还是过多,无法直接作为transformer的输入,作者想到了利用1维卷积把数据特征降低,然后再送到transformer里。个人觉得没什么道理。另外如题目所说,Transformer后的输出就是简单的分类,并没有像上一篇一样进行重建训练,这也导致了这个模型没有任务泛化能力。
整体的模型结构比较好懂,如下图所示:
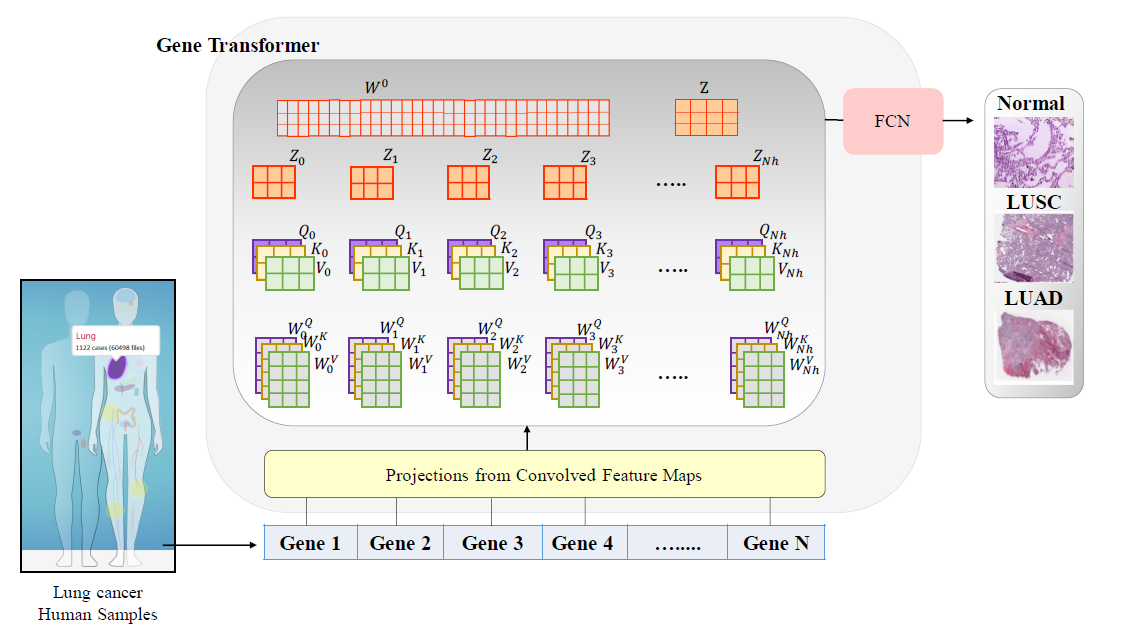
结果自然是和很多方法都进行了比较,发现效果都很不错,有些F1 score达到了1,不知道是不是反向说明任务太简单了或者模型过拟合了。
都看到这里了,点个赞再走吧

文章评论